Qualitatively, entropy is simple. What it is, why it is so useful in understanding the behavior of macro systems or of molecular systems is easy to state. The key to simplicity has been mentioned by chemists and physicists for a century and a half, but not developed in detail.1 In classical thermodynamics, it is viewing entropy increase as a measure of the dispersal of energy from localized to spread out at a given temperature (as in the examples of the next section). In "molecular thermodynamics" it is considering the change in a system (or surroundings or their total) for which there are fewer accessible microstates to one for which there is an increased number of accessible microstates (a delocalization of energy, as described in later sections). Most important to educators, emphasizing energy dispersal even qualitatively can introduce a direct relationship of molecular behavior to entropy change, an idea that can be readily understood by beginning college students.
This view was broached partially in a previous article that showed "disorder" was outmoded and misleading as a descriptor for entropy (1, 2). The present article substantiates the power of seeing entropy change as measuring various modes of energy dispersal (in part, by Leff [3a]) in classical and molecular thermodynamics in important equilibrium situations and chemical processes.
The preceding introduction in no way denies the subtlety or the difficulty of many aspects of thermodynamics involving entropy. In numerous cases the theory or calculation or experimental determination of entropy change can be overwhelming even to graduate students and challenging to experts. In complex cases, the qualitative relation of energy dispersal to entropy change can be so inextricably obscured that it is moot. However, this does not weaken its explanatory power in the common thermodynamic examples presented to beginning students, nor in using energy dispersal to introduce them to spontaneous change in chemistry.
A hot pan spontaneously disperses some of its energy to the cooler air of a room. Conversely, even a cool room would disperse a portion of its energy to colder ice cubes placed in it. When a container of nitroglycerine is merely dropped on the floor, the nitroglycerine may change into other substances explosively, because some of its bond energy is spread out in increasing the vigorous motions of molecules in the gaseous products. At a high pressure in a car tire, the compressed air tends to blow out and disperse its more concentrated energy to the lower pressure atmosphere. At any pressure, ideal gases will spontaneously flow into an evacuated chamber, spreading the energy of their molecular motions over the final larger volume without any change in temperature. Batteries, small or large, “lose their charge” (spontaneously reacting to decrease the amount of the reactants) and thereby disperse their chemical potential energy in heat if even a slightly conductive path is present between the two poles. . Hydrogen and oxygen in a closed chamber will remain unaltered for years and probably for millennia, despite their larger combined bond energies compared to water. However, if a spark is introduced, they will react explosively — a phenomenon characteristic of rapid reactions in which a portion of the reactants' bond energies disperses in causing a very great increase in the product molecules' motion that ultimately becomes spread out in molecular motional energy in the surroundings.
In these seven varied examples, and in all everyday spontaneous physical happenings and chemical reactions, some type of energy flows from being localized or concentrated to becoming spread out to a larger space, always to a state with a greater number of microstates. This summary will be given precise descriptions in macro thermodynamics and in molecular behavior later.
Most spontaneous chemical reactions are exothermic. The products that are formed have lower bond energies than the starting materials. Thus, as a result of such a reaction, some of the reactants' bond energy is dispersed to cause increased molecular motion in the products and thence, if the system is not isolated, to cooler surroundings. Endothermic reactions are less common at room temperatures (but routine at blast furnace temperatures). In this type of reaction, substances (alone or with others) react because energy from the more-concentrated-energy surroundings is dispersed to the bonds and motions of the molecules of the less-concentrated-energy substances in a system. The result is the formation of new substances with larger bond energies than their starting materials.
Parallel with the behavior of energy in chemical phenomena is its dispersal in all spontaneous physical events, whether they are complex and exotic, or as simple and too common as what happens to the kinetic energy of a car in a collision.
Clausius' definition of entropy change, dS = Dqrev/T, could be expressed verbally as "entropy change = the amount of energy dispersed reversibly at a specific temperature T". Clausius' synonym for entropy was Verwandlung, transformation. Any definition needs exposition, but certainly this definition does not imply that entropy is "disorder" nor that it is a measure of "disorder" nor that entropy is a driving force. Instead, the reason for transformation in classical thermodynamics is energy's dispersing from a source that is almost imperceptibly above T to a receptor that is at T, or spreading out from where it is confined in a small space to a larger volume whenever it is not restricted from doing so, or dispersing throughout a mixture of fluids or solid solute and liquid. The classical (macro) thermodynamic explanation of entropy in Clausius' equation lies in its measuring or indexing the process of energy dispersal by qrev/T.
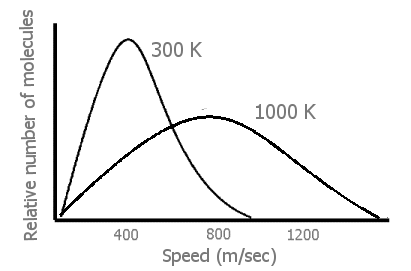
Early in their discussion of kinetic-molecular theory, most general chemistry texts have a Figure of the greatly increased distribution of molecular speeds at higher temperatures in gases than at moderate temperatures.
Figure 1: Generalized plot of molecules at 300 K vs. 1000K (100m/sec = 224mph) Heavier molecules have broader peak s at both temperatures.
When the temperature of
a gas is raised (by transfer of energy from the surroundings of the system),
there is a great increase in the velocity, v, of many of the gas molecules
(Figure 1). From 1/2mv2, this means
that there has also been a great increase in the
translational energies of
those faster moving molecules. Finally, we can see that an input of energy
not only causes the gas molecules in the system to move faster — but also
to move at very many different fast speeds. (Thus, the energy in a
heated system is more dispersed, spread out in being in many separate speeds
rather than more localized in fewer moderate speeds.)
A symbolic indication of the different distributions of the translational
energy of each molecule of a gas on low to high energy levels in a 36-molecule
system is in Figure 2, with the lower temperature gas as 2A and the higher
temperature gas as 2B.
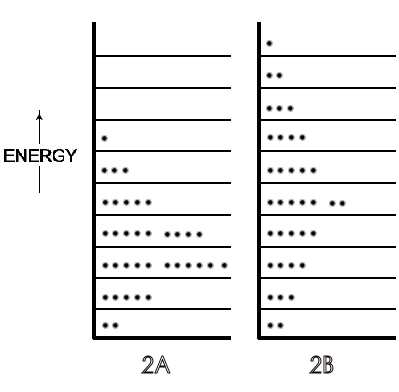
Figure 2: [A] is a distribution of gas molecules on specific energy levels at 300 K and [B] is the same group of gas molecules on energy levels at 1000 K.
These and later Figures in this section are symbolic because, in actuality, this small number of molecules is not enough to exhibit thermodynamic temperature. For further simplification, rotational energies that range from zero in monatomic molecules to about half the total translational energy of di- and tri-atomic molecules (and more for most polyatomic) at 300 K are not shown in the Figures. If those rotational energies were included, they would constitute a set of energy levels (corresponding to a spacing of ~10-23 J) each with translational energy distributions of the 36 molecules (corresponding to a spacing of ~10-37 J). These numbers show why translational levels, though quantized, are considered virtually continuous compared to the separation of rotational energies. The details of vibrational energy levels — two at moderate temperatures (on the ground state of which would be almost all the rotational and translational levels populated by the molecules of a symbolic or real system) — can also be postponed until physical chemistry. At this point in the first year course, depending on the instructor's preference, only a verbal description of rotational and vibrational motions and energy level spacing need be introduced.
By the time in the beginning course that students reach thermodynamics, five to fifteen chapters later than kinetic theory, they can accept the concept that the total motional energies of molecules includes not just translational but also rotational and vibrational movements (that can be sketched simply as in Figure 1 of 2ndlaw.com/entropy.html). This total motional energy— just as translational and all other kinds of energy — is distributed (quantized) on specific energy levels. Figure 2A and B can be viewed as prototypes of more complex diagrams of molecular energies on specific levels, with additional higher energy levels populated (as well as prior higher energy levels more populated) when the temperature of a system is raised. Further, students can sense that, at the same temperature as in gases, molecules in liquids move, and rotate, and vibrate internally with the same total energy, but just do not travel as far before colliding.
A microstate is one of many arrangements of the molecular energies (i.e., ‘the molecules on each particular energy level') for the total energy of a system. Thus, Figure 2A is one microstate for a system with a given energy and Figure 2B is a microstate of the same system but with a greater total energy. Figure 3A (just a repeat of 2A, for convenience) is a different microstate than the microstate for the same system shown in 3B; the total energy is the same in 3A and 3B but in 3B the arrangement of energies has been changed because two molecules have changed their energy levels, as indicated by the arrows.
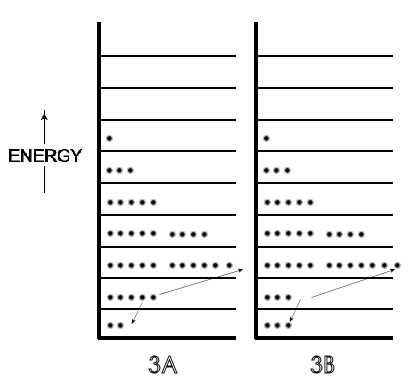
Figure 3: [A] is the same as [2A], one microstate of molecules at 300 K. [B] is a different microstate than 2[A] of the same system of 300 K molecules — the same total energy but two molecules in [B] having different energies than they had in the arrangement of 2[A], as indicated by the arrows.
A possible scenario for that different microstate in Figure 3 is that these two molecules on the second energy level collided at a glancing angle such that one gained enough energy to be on the third energy level, while the other molecule lost the same amount of energy and dropped down to the lowest energy level. In the light of that result of a single collision and the billions of collisions of molecules per second in any system at room temperature, there can be a very large number of microstates even for this system of just 36 molecules in Figures 2 and 3. (This is true despite the fact that not every collision would change the energy of the two molecules involved, and thus not change the numbers on a given energy level. Glancing collisions could occur with no change in the energy of either participant.) For any real system involving 6 x 1023 molecules, however, the number of microstates becomes humanly incomprehensible for any system, even though we can express it in numbers, as will now be developed.
The quantitative entropy change in a reversible process is given by ΔS = qrev /T. (Irreversible processes involving temperature or volume change or mixing can be treated by calculations from incremental steps that are reversible.) According to the Boltzmann entropy relationship, ΔS = kB ln WFinal / WInitial, where kB is Boltzmann's constant and WFinal or WInitial is the count of how many microstates correspond to the Final or Initial macrostates. The number of microstates for a system determines the number of ways in any one of which that the total energy of a macrostate can be at one instant. Thus, an increase in entropy means a greater number of microstates for the Final state than for the Initial. In turn, this means that there are more choices for the arrangement of a system's total energy at any one instant, far less possibility of localization (such as cycling back and forth between just 2 microstates), i.e., greater dispersal of the total energy of a system because of so many possibilities.
Some instructors may prefer “delocalization” to describe the status of the
total energy of a system when there are a greater number of microstates rather
than fewer, as an exact synonym for “dispersal” of energy as used here in
this article for other situations in chemical thermodynamics. The advantage
of uniform use of ‘dispersal' is its correct common-meaning applicability
to examples ranging from motional energy becoming literally spread out in
a larger volume to the cases of thermal energy transfer from hot surroundings
to a cooler system, as well as to distributions of molecular energies on
energy levels for either of those general cases. Students of lesser ability
should be able to grasp what ‘dispersal' means in three dimensions, even
though the next steps of abstraction to what it means in energy levels and
numbers of microstates may result in more of a ‘feeling' than a preparation
for physical chemistry that it can be for the more able.
Of course, dispersal of the energy of a system in terms of microstates does not mean
that the energy is smeared or spread out over microstates like peanut butter
on bread! All the energy of the macrostate is always in only one microstate
at one instant. It is the possibility that the total energy of the macrostate
can be in any one of so many more different arrangements
of that energy at the next instant — an increased probability that
it could not be localized by returning to the same microstate — that
amounts to a greater dispersal or spreading out of energy when there are
a larger number of microstates
(The numbers of microstates for chemical systems above 0 K are astounding.
For any substance at a temperature about 1-4 K, there are
Summarizing, when a substance is heated, its entropy increases because the energy acquired and that previously within it can be far more dispersed on the previous higher energy levels and on those additional high energy levels that now can be occupied. This in turn means that there are many many more possible arrangements of the molecular energies on their energy levels than before and thus, there is a great increase in accessible microstates for the system at higher temperatures. A concise statement would be that when a system is heated, there are many more microstates accessible and this amounts to greater delocalization or dispersal of its total energy. (The common comment "heating causes or favors molecular disorder" is an anthropomorphic labeling of molecular behavior that has more flaws than utility. There is virtual chaos, so far as the distribution of energy for a system (its number of microstates) is concerned, before as well as after heating at any temperature above 0 K and energy distribution is at the heart of the meaning of entropy and entropy change. ) (5).
When the volume of a gas is increased by isothermal expansion into an evacuated
container, an entropy increase occurs but not for the same reason as when
a gas or other substance is heated. There is no change in the quantity
of energy of the system in such an expansion of the gas; dq is zero. Instead,
there is a spontaneous dispersal or spreading out of that energy in space.
This change in entropy can be calculated in macrothermodynamics from the
equivalent qrev to the work required to reversibly compress a
mole of the gas back to its original volume, i.e., RT ln (V2 /V1 ),
and then ΔS = R
ln (V2 /V1 ). From the viewpoint of molecular thermodynamics,
a few general chemistry texts use quantum mechanics to show that when a gas
expands in volume, its energy levels become closer together in any small
range of energy.
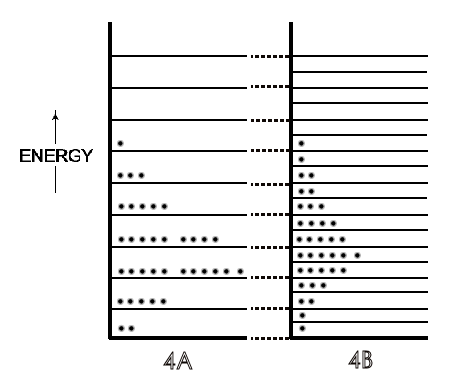
Figure 4: [A] is the system of molecular energies ('of molecules') on particular energy levels as in 2[A] and 3[A]. [B] is the [A] system after its volume has doubled. There are two energy levels for every energy level in 4[A] due to the increase in volume. The total energy has become dispersed over many more energy levels than in 4[A].
Symbolically in Figure 4B, a doubling of volume doubles the number of energy
levels and increases the possibilities for energy dispersal because of these
additional levels for the same molecular energies in 4A. Due to this increased
possibility for energy dispersal — a spread over twice as many energy levels
— the entropy of the system increases. Then, as could be expected from
any changes in the population of energy levels for a system, there are also
far greater numbers of possible arrangements of the molecular energies on
those additional levels, and thus many more microstates for the system. This
is the ultimate quantitative measure of an entropy increase in molecular
thermodynamics, a greater kB ln WFinal / WInitial .
When two dissimilar ideal gases mix and the volume increases, or when dissimilar liquids mix with or without a volume change, the number of energy levels that can be occupied by the molecules of each component increases. Thus, for somewhat different reasons there are similar results in this progression: additional energy levels for population by molecules (or ‘by molecular energies'), increased possibilities for motional energy dispersal on those energy levels, a far greater number of different arrangements of the molecules' energies on the energy levels, and the final result of many more accessible microstates for the system. There is an increase in entropy in the mixture. This is also the case when solutes of any type dissolve (mix) in a solvent. The entropy of the solvent increases (as does that of the solute). This phenomenon is especially important because it is the basis of colligative effects that will be discussed later.
The entropy of a substance at any temperature T is not complex or mysterious. It
is simply a measure of the total amount of energy that had to be dispersed
within the substance (from the surroundings) from 0 K to T, incrementally and
reversibly and divided by T for each increment, so the substance could exist
as a solid or (with additional reversible energy input for breaking intermolecular
bonds in phase changes) as a liquid or as a gas at the designated temperature. Because
the heat capacity at a given temperature is the energy dispersed in a substance
per unit temperature, integration from 0 K to T of Cp/T
dT (+ q/T for any phase change) yields the total entropy. This result, of
course, is equivalent to the area under the curve to T in Figure 5.
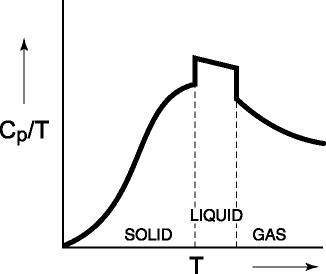
Figure 5: The Cp/T
vs. T from 0 K for a substance. The area under the curve (plus q/T at any
phase change) is the entropy of the substance at T.
© Journal of Chemical Education.
To change a solid to a liquid at its melting point requires large amounts of energy to be dispersed from the warmer surroundings to the solid for breaking the intermolecular bonds to the degree required for existence of the liquid at the fusion temperature. (“To the degree required” has special significance in the melting of ice. Many, but not all of the hydrogen bonds in crystalline ice are broken. The rigid tetrahedral structure is no longer present in liquid water but the presence of a large number of hydrogen bonds is shown by the greater density of water than ice due to the even more compact hydrogen-bonded clusters of H2O.) Quantitatively, the entropy increase in this isothermal dispersal of energy from the surroundings is ΔHFusion /T. Because melting involves bond-breaking, it is an entropy increase in the potential energy of the substance involved. (This potential energy remains unchanged in a substance throughout heating, expansion, mixing, subsequent phase change to a vapor, or mixing. Of course, it is released when the temperature of the system drops below the freezing/melting point).The process is isothermal, and therefore there is no energy transferred to the system to increase motional energy. However, a change in the motional energy — not an increase in the quantity of energy — occurs from the transfer of vibrational energy in the ice crystal to the liquid. When the liquid forms, there is rapid breaking of hydrogen bonds (trillionths of a second) and forming new ones with adjacent molecules. This might be compared to a fantastically huge dance in which the individual participants don't move very far (takes a water molecule >12 hours to move a cm at 298K) but they are holding hands and then releasing to grab new partners far more frequently than billions of times a second. Thus, that previous motional energy of intermolecular vibration that was in the crystal is now distributed among a far greater number of new translational energy levels, and that means that there are many more accessible microstates than in the solid. Similarly, a liquid at its vaporization temperature has the same energy as its gas molecules. (All of the enthalpy of vaporization is needed to break intermolecular bonds in the liquid.) However in the case of liquid to vapor, there is a huge expansion (a thousand times increase)in volume. Therefore, this means closer energy levels, far more than were available for the motional energy in the liquid — and a greatly increased number of microstates for the vapor.
The entropy effects in gas expansion into a vacuum, as described previously, are qualitatively similar to gases mixing.. From a macro viewpoint, the initial energy of each constituent becomes more dispersed in the new larger volume provided by the combined volumes of the components. Then, on a molecular basis, because the density (closeness) of energy levels increases in the larger volume, and therefore there is greater dispersal of the molecular energies on those additional levels, there are more possible arrangements (more microstates) for the mixture than for the individual constituents. Thus, the mixing process for gases is actually a spontaneous process due to an increase in volume. The entropy increases (6). Meyer vigorously pointed toward this same statement in "the spontaneity of processes generally described as "mixing", that is, combination of two different gases initially each at pressure, p, and finally at total pressure, p , has absolutely nothing to do with the mixing itself of either" (7).
The same cause — of volume increase of the system resulting in a greater density of energy levels — obviously cannot apply to liquids in which there is little or no volume increase when they are mixed. However, as Craig has well said, "The "entropy of mixing" might better be called the "entropy of dilution" (8b). By this one could mean that the molecules of either constitutent in the mixture become to some degree separated from one another and thus their energy levels become closer together by an amount determined by the amount of the other constituent that is added. Whether or not this is true, ‘configurational entropy' is the designation in statistical mechanics for considering entropy change when two or more substances are mixed to form a solution. The model uses combinatorial methods to determine the number of possible “cells” (that are identified as microstates, and thus each must correspond to one accessible arrangement of the energies of the substances involved). This number is shown to depend on the mole fractions of each component in the final solution and thus the entropy is found to be: ΔS = - R (n1 ln X1 + n2 ln X2 ) with n1 and n2 the moles of pure solute and solvent, and X1 and X2 the mole fractions of solute and solvent (8a, 9).
In general chemistry texts, configurational entropy is called ‘positional entropy' and is contrasted to the classic entropy of Clausius that is then called ‘thermal entropy'. The definition of Clausius is fundamental; positional entropy is derivative in that its conclusions can be derived from thermal entropy concepts/procedures, but the reverse is not possible. Most important is the fact that positional entropy in texts often is treated as just that: the positions of molecules in space determine the entropy of the system, as though their locations — totally divorced from any motion or any energy considerations — were causal in entropy change. This is misleading. Any count of ‘positions in space' or of ‘cells' implicitly includes the fact that molecules being counted are particles with energy. Although the initial energy of a system is unchanged when it increases in volume or when constituents are mixed to form it, that energy is more dispersed, less localized after the processes of expansion or of mixing. Entropy increase always involves an increase in energy dispersal at a specific temperature.
"Escaping tendency" or chemical potentials or graphs that are complex to a beginner are often used to explain the freezing point depression and boiling point elevation of solutions. These topics can be far more clearly explained by first describing that an entropy increase occurs when a non-volatile solute is added to a solvent — the solvent's motional energy becomes more dispersed compared to the pure solvent, just as it does when any non-identical liquids are mixed. (This is the fundamental basis for a solvent's decreased "escaping tendency" when it is in a solution. If the motional energy of the solvent in a solution is less localized, more spread out, the solvent less tends to “escape” from the liquid state to become a solid when cooled or a vapor when heated.)
Considering the most common example of aqueous solutions of salts: Because of its greater entropy in a solution (i.e., its energy more ‘spread out' at 273.15 K and less tending to have its molecules ‘line up' and give out that energy in forming bonds of solid ice), liquid water containing a solute that is insoluble in ice is not ready for equilibrium with solid ice at 273.15 K. Somehow, the more-dispersed energy in the water of the solution must be decreased for the water to change to ice. But that is easy, conceptually — all that has to be done is to cool the solution below 273 K because, contrary to making molecules move move rapidly and spread their energy when heated, cooling a pure liquid or a solution obviously will make them move more slowly and their motional energy become less spread out, more like the energy in crystalline ice. Interestingly, the heat capacity of water of an aqueous solution (75 J/mol) is about twice that of ice (38 J/mol). That means that when the temperature of the surroundings decrease by one degree, the solution disperses much more energy to the surroundings than the ice — and the motional energy of water and any possible ice becomes closer. Therefore, when the temperature decreases a degree or so (and finally to –1.86 C/kg.mol!), the solution's higher entropy has rapidly decreased to be in thermodynamic equilibium with ice and the surroundings amd freezing can begin. As energy (the enthalpy of fusion) continues to be dispersed to the cold surroundings, the liquid water freezes.
Of course, the preceding explanation could have been framed in terms of entropy change or numbers of microstates, but keeping the focus on what is happening to molecular motion, on energy and its dispersal is primary rather than derivative.
The elevation of boiling points of solutions having a non-volatile solute is as readily rationalized as is freezing point depression. The more dispersed energy (and greater entropy) of a solvent in the solution means that the expected thermodynamic equilibrium (the equivalence of the solvent's vapor pressure at the normal boiling point with atmospheric pressure) cannot occur at that boiling point. For example, the energy in water in a solution at 373 K is more widely dispersed due to the increased number of microstates for a solution than for pure water at 373 K. Therefore, water molecules in a solution less tend to leave the solution for the vapor phase than from pure water. Energy must be transferred from the surroundings to an aqueous solution to increase its energy content, thereby compensating for the greater dispersion of the water's energy due to its being in a solution, and to allow water molecules to escape readily to the surroundings. (More academically, “to raise the vapor pressure of the water in the solution”.) As energy is dispersed to the solution from the surroundings and the temperature rises above 373 K, at some point a new equilibrium temperature for phase transition is reached. The water then boils because the greater vapor pressure of the water in the solution with its increased motional energy now equals the atmospheric pressure.
Although the hardware system of osmosis in the chemistry laboratory and in industry is unique, the process that occurs in it is merely a special case of the mixing of a solvent with a solution — ‘special' because of the existence of such marvels as semi-permeable membranes through which a solvent can pass but a solute cannot. As would be deduced from the discussion about mixing two liquids or mixing a solid solute and a liquid, the solvent in a solution has a greater entropy than a sample of pure solvent. Its energy is more dispersed in the solution. Thus, if there is a semi-permeable membrane between a solution made with a particular solvent and some pure solvent, the solvent will spontaneously move through the membrane to the solution because its energy becomes more dispersed in the solution and thus, its entropy becomes increased if it mixes with the solution.
The following section discusses a general view of spontaneous chemical reactions wherein energy changes occur in chemical bonds that disperse energy in the surroundings.
Undoubtedly, the first-year chemistry texts that directly state the Gibbs equation as ΔG = ΔH - T ΔS, without its derivation involving the entropy of the universe, do so in order to save valuable page space. However, such an omission seriously weakens students' understanding that the "free energy" of Gibbs is more closely related to entropy than it is to energy (10). This quality of being a very different variety of energy is seen most clearly if it is derived as the result of a chemical reaction as is shown in some superior texts, and developed below.
If there are no other events in the universe at a particular moment that a spontaneous chemical reaction occurs in a system, the changes in entropy are: ΔSuniverse = ΔSsurroundings + ΔSsystem . However, the internal energy evolved from changes in bond energy, -ΔH, in a spontaneous reaction in the system passes to the surroundings. This - ΔHsystem, divided by T, is ΔSsurroundings Inserting this result (of - (ΔH/T)system = ΔSsurroundings) in the original equation results in ΔSuniverse = - (ΔH/T)system + ΔSsystem. But, the only increase in entropy that occurred in the universe initially was the dispersal of differences in bond energies as a result of a spontaneous chemical reaction in the system. To that dispersed, evolved bond energy (that therefore has a negative sign), we can arbitrarily assign any symbol we wish, say - ΔG in honor of Gibbs, which, divided by T becomes ( - ΔG/T)system. Inserted in the original equation as a replacement for ΔSuniverse (because it represents the total change in entropy in the universe at that instant): - (ΔG/T)system = -(ΔH/T)system + ΔSsystem. Finally, multiplying the whole by -T, we obtain the familiar Gibbs equation, ΔG = ΔH - TΔS, all terms referring to what happened or could happen in the system.
Strong and Halliwell rightly maintained that - ΔG, the "free energy", is not a true energy because it is not conserved (10). Explicitly ascribed in the derivation of the preceding paragraph (or implicitly in similar derivations), - ΔG is plainly the quantity of energy that can be dispersed to the universe, the kind of entity always associated with entropy increase, and not simply energy in one of its many forms. Therefore, it is not difficult to see that ΔG is indeed not a true energy. Instead, as dispersible energy, when divided by T, ΔG/T is an entropy function — the total entropy change associated with a reaction, not simply the entropy change in a reaction, i.e., Sproducts - Sreactants, that is the ΔSsystem. This is why ΔG has enormous power and generality of application, from prediction of the direction of a chemical reaction, to prediction of the maximum useful work that can be obtained from it. The Planck function, - ΔG/T, is superior in characterizing the temperature dependence of the spontaneity of chemical reactions (11, 12). These advantages are the result of the strength of the whole concept of entropy itself and the relation of ΔG to it, not a result of ΔG being some novel variant of energy.
A significant contribution to teaching is using entropy to explain quickly and simply why many chemical reactions do not go to completion. ("Completion" is here used in the sense that no reactants remain after the reaction; products are formed in 100% yield.) Why shouldn't a reaction go ‘all the way to the right' as written? Many texts say something like "systems can achieve the lowest possible free energy by going to equilibrium, rather than proceeding to completion" and well support the statement by quantitative illustrations of the free energies of forward and reverse reactions. A number of texts illustrate the failure of many reactions to go to completion with curves of the progress of a reaction. Few texts present a qualitative description of the entropy changes (which are essentially ΔG/T changes) that immediately simplify the picture for students.
As mentioned repeatedly in this article, mixtures of many types have higher entropy than their pure components (that is not caused by any profound or novel "entropy of mixing" but is simply due to the greater number of microstates for a system that is a mixture compared to the number of microstates for the total of the separate pure components. ) Thus, students who have been told about this mixing phenomenon can immediately understand why, in a reaction of A to make B, the entropy of a mixture of B with a small residual quantity of A would be greater than that of pure B. From this, students can see that the products in many such reactions would more probably be such a mixture because this would result in a greater entropy increase for the overall reaction than formation of only pure B. The qualitative conclusion that reactions that are moderately exothermic go to an equilibrium of B plus some A rather than to completion readily becomes apparent on this basis of maximal entropy change, determined by - R ln K, (DGo/T).3
Energy of all types changes from being localized to becoming dispersed or spread out, if the process is not hindered. Most frequently in chemistry, it is the motional energy of molecules or bond energies that is dispersed in the system or the surroundings so that there is a larger number of microstates than initially.
This statement of the second law is not complete without identifying the overall process as an increase in entropy including the fact that spontaneous thermodynamic entropy change has two requisites: The motional energy of molecules that most often enables entropy change is only actualized if the process makes available an increased number of microstates, a probability requisite. Thereby, thermodynamic entropy change is clearly distinguished from information "entropy" by having two essential requisites, energy as well as probability. (Information "entropy" has only one requisite, probability.)
The increase of entropy, identifiable most readily as the dispersal of energy at a temperature, gives direction to physical and chemical events. Energy's dispersing is probabilistic rather than deterministic. As a consequence, there may be small sections of a larger system in which energy is temporarily concentrated. (Brownian motion is an example of near-microscopic deviation. Chemical substances having greater free energies than their possible products are common examples. They continue to exist because of activation energies that act as barriers to immediate chemical reaction that would form lower energy substances resulting from the dispersion of portions of their bond energies. “Chemical kinetics firmly restrains time's arrow in the taut bow of thermodynamics for milliseconds or for millennia.”) (13).
Entropy change is measured by the reversible dispersion of energy at a temperature T. Entropy is not disorder, not a measure of disorder, not a driving force. The entropy of a substance, its entropy change from 0 K to any T, is a measure of the energy that has been incrementally (reversibly) dispersed from the surroundings to the substance at the T of each increment, i.e., integration from 0 K to T of Cp/T dT (+ q/T for any phase change).
A molecular view of entropy is most helpful in understanding entropy change. Microstates are quantum mechanical descriptions of ways that molecules can differ in their energy distributions in a macro system; an enormous number of microstates are possible for any macrostate above 0 K (5). When a macrostate is warmed, a greater dispersal of energy occurs in a substance or a system because many additional microstates become accessible. This increased spreading out of energy is reflected in the entropy increase by Cp/T dT. . Liquids have greater numbers of microstates than their solid phase at the melting point equilibrium because the cleavage of intermolecular bonds in the solid, indicated by the enthalpy of phase change, a potential energy change in the system. This cleavage allows increased translational motion in the liquid from molecular movement while rapidly breaking and making new hydrogen bonds (the energy coming from the solid's crystal vibration modes). The many additional energy levels result in an enormous increase in possible arrangements of the molecular energies and thus many more microstates for the liquid than were accessible for the solid. Further great increases in accessible microstates become actualized for comparable reasons in the change to a vapor phase. Thus, greater dispersal of energy is indicated by an increase in entropy due to phase change from 0 K to T.
Gas expansion into a vacuum, mixing of ideal gases or liquids, diffusion, effusion, colligative effects, and osmosis each fundamentally involves an increase in entropy due to increased dispersion of energy (with no change in total energy in the case of ideal substances) due to a greater number of microstates for the particular system involved.
At its heart, the Gibbs equation is an "all-entropy" equation of state. The Gibbs energy has been accurately criticized as not "a true energy". It is not conserved. The Gibbs energy, - ΔG, can be correctly described as "dispersible energy" rather than a variety of energy, because of its ultimate definition as the energy portion of an entropy function, - ΔG/T. Modestly exothermic reactions go to equilibrium rather than to completion because a mixture of products with some reactant has a larger entropy than pure products alone. Highly exothermic reactions proceed essentially to completion because the enthalpy transfer to the surroundings constitutes an overwhelmingly greater entropy change in the total of [system + surroundings] than any small counter from the entropy change due to mixing of products with unaltered reactants in the system.
I thank Norman C. Craig for his invaluable detailed critiques of this article and the great number of corrections to which he has led me since 2000. There were many remaining flaws in the original ms. and publication that were all my errors. I trust that they have been corrected in this 2005 revision but will appreciate further corrections by readers. Harvey S. Leff, Ralph Baierlein, and Walter T. Grandy, Jr. answered many questions in the development of the 2002 ms. and Harvey Leff has continued to do so.
Chemical "bond energies" are examples of potential energy. Potential energy — whether in a physical object like a ball held high or in a chemical substance's bond energy as in TNT — does not spontaneously spread out. It can't because it is hindered from doing so. Only when potential energy is changed into kinetic energy — by letting the ball fall or by impacting the TNT with a blasting cap — does it begin to be dispersed. As Professor W. B. Jensen of the University of Cincinnati has emphasized [ J. Chem.Educ. 2004 , 639-640], it is really kinetic energy that disperses or spreads out in space from a source that we would say had a great deal of potential energy.
Then, it is primarily the dispersal of "motional energy", the kinetic energy of translation (movement in space), or rotation, or vibration (of atoms inside their molecules) and vibration of particles in crystals, that entropy measures as a function of temperature. (The potential energy changes that occur in phase changes are due to breaking or forming relatively weak inter molecular bonds. They too are included in any total figure for entropy values but PE phase change energies are not altered in many cases that are discussed in this article: heating (at any temperature other than phase change temps!), gas expansion, fluid or solid-liquid mixing. Of course, they may be involved in chemical reactions.)
Before focusing on chemical systems for a class of beginners, the instructor can profitably illustrate the spontaneous dispersal of energy as a common phenomenon in everyday happenings. They could range from a bouncing ball ceasing to bounce, to the results of a car crash (KE dispersed to twisted warm metal, etc.), to highly charged clouds (yielding lightning bolts and the heated air creating thunder , to attenuation effects with which students are familiar (from the just-mentioned thunder, to loud musical sounds a room or dorm away) or ripple heights (from an object thrown in a swimming pool). All forms of energy associated with common objects (and caused by energy input separate from them) as well as with ( innately energetic ) molecules become more dissipated, spread out, dispersed if there are ways in which the energy can become less localized and the dispersal process is not hindered. (In the case of molecules where energy must be considered quantized, these ways of becoming less localized are described as any process that yields an increased number of accessible microstates.)